INTRODUÇÃO
Uma dúvida que paira entre os assuntos relativos à esterilização de produtos para saúde é: Um produto esterilizado em uma embalagem de papel grau cirúrgico com filme laminado pelo processo de esterilização por vapor saturado, tem a mesma validade de esterilização de um produto esterilizado no mesmo tipo de embalagem em óxido de etileno?
Resposta : SIM e NÃO
SIM: No tocante ao método de esterilização propriamente dito, tanto faz um material ter sido esterilizado em vapor saturado, quanto um outro ter sido esterilizado em óxido de etileno. Se a população inicial de micro organismos atingiu um SAL (nível de segurança aceitável definido pela Norma NBR ISO 17665-1) de 10-6, é seguro afirmar que haverá no máximo um micro organismo viável em um milhão de amostragens, portanto ambos materiais são considerados igualmente estéreis. Em outras palavras, não é exatamente o método de esterilização que define maior ou menor garantia, pois o ponto de partida, produto estéril, é condição igual nos dois processos.
NÃO: Considerando que a manutenção da esterilidade não está associada ao tipo de esterilização, mas à manutenção dessa condição, porque então produtos esterilizados em ETO e Vapor saturado têm prazos comumente tão distintos?
Sabendo-se que a determinação do prazo de validade não está associada ao tempo e sim ao evento relacionado à quebra da barreira bacteriana e perda de esterilidade, torna-se imperativo a análise dos materiais que mantêm o produto livre de micro-organismos estéreis em condições de armazenagem, ou seja, A EMBALAGEM.
MANUTENÇÃO DA ESTERILIZAÇÃO
As informações dessa matéria foram obtidas dos fabricantes de papel grau cirúrgico. O papel utilizado na embalagem de produtos para saúde é um dos fatores que contribuem para minimizar o risco de contaminação e aumentar a segurança dos pacientes. Entre a data da esterilização e a data de utilização dos artigos esterilizados a embalagem primária formada por papel grau cirúrgico e filme laminado é a principal barreira contra o risco externo de contaminação. Atualmente Normas como as NBRs ISO 14990 e 11607 não tratam mais de “embalagens de esterilização” mas sim de “sistemas de barreira estéril” sob o conceito que a esterilidade não é resultado somente da embalagem primária, mas do conjunto como um todo que envolve a embalagem secundária e provavelmente até mesmo uma terceira, porém é indiscutível a função fundamental da embalagem com papel grau cirúrgico, na manutenção da esterilidade.
CARACTERÍSTICAS DOS PAPEIS PARA ESTERILIZAÇAO
Estes papeis são desenvolvidos afim de promover a selagem do filme laminado à uma de suas faces, a passagem do agente esterilizante para esterilização e a manutenção da barreira bacteriana resistindo ao transporte e à armazenagem. Outras propriedades como aceitar o processo de impressão e permitir abertura asséptica também são importantes, porém serão abordadas em outras matérias.
Afim de esclarecer sobre o prazo de validade determinado pelas materiais primas é importante entender duas características cujos nomes muito se confundem entre os usuários são elas: porosidade e diâmetro de poros.
A porosidade, também conhecida como permeância, é a propriedade que determina a de passagem do agente esterilizante durante um determinado período. Em outras palavras é o que determina a facilidade do agente esterilizante entrar e sair pelo papel. Essa propriedade está relacionada à forma como as fibras do papel estão dispostas e à concentração de poros por unidade de área.
O diâmetro de poros é a propriedade que determina a capacidade do material em filtrar os agentes externos. Maior será a barreira quanto menores forem os poros.
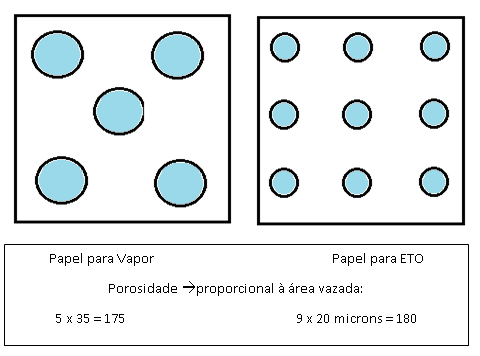
As Normas NBR 14990 e EN 868 determinam que o diâmetro máximo de poros para papeis de ETO é 20 microns na média e para papeis de vapor o diâmetro máximo é 35 microns na média.
Isso significa que nos exemplos hipotéticos abaixo, se tivermos no vapor um papel com poros de 35 micros e no ETO poros de 20 microns a porosidade em ambos é praticamente igual, porém a barreira bacteriana do caso 2 é muito maior, já que os poros são menores e dificultam mais a passagem dos micro organismos ou seja, garantia de esterilidade superior.
Há um ponto de equilíbrio pretendido pelos fabricantes de papel. Permitir a maior porosidade, com a melhor barreira e manter a maior resistência mecânica. Normalmente ao se fabricar um papel, a quantidade de poros por unidade de área, que determina a permeância ao agente esterilizante, é aumentada ao máximo até o limite em que o papel atinge a resistência mecânica mínima permitida pelas Normas.
EM RESUMO
Papel para vapor: Possui porosidade capaz de permitir a passagem do vapor e mais fácil ainda do ETO, diâmetro de poros máximo 35 microns, porém com “shelf life” (tempo de vida) para esterilidade de no máximo 6 meses em condições normais de utilização dentro do ambiente controlado dos arsenais de Centrais de Esterilização.
Papel para ETO : Possui porosidade capaz de permitir a passagem do ETO porém muito fechado para a passagem do vapor. Diâmetro de poros máximo 20 microns. Seu uso em processos de esterilização em vapor é muito complicado, pois dificulta muito a secagem, sem contar a possibilidade de rompimento da embalagem nas fases de vácuo das autoclaves. Devida a alta barreira oferecida pelo diâmetro dos poros, o “shelf life” de esterilização é normalmente definido pelos fabricantes de material médico superior a 5 anos, o que permite para as empresas garantir uma vida útil equivalente de seus produtos.
Normalmente os fabricantes de produtos médicos no BRASIL, não se importam em qualificar seus papeis para uso adequado e acabam utilizando os papeis desenvolvidos para VAPOR para embalar produtos que são tipicamente esterilizados em ETO. Da mesma forma os convertedores de embalagens, que fabricam embalagem para ETO, não alertam seus clientes fabricantes de produtos médicos, que poderiam utilizar um papel específico para ETO.
Papel de uso misto: Na verdade esses papeis não existem. É impossível com as tecnologias atuais satisfazer o diâmetro de poros máximo do ETO, a permeância mínima do vapor e ainda a resistência mecânica de ambos os processos. Os papeis oferecidos pelas industrias aos hospitais, que contém os dois indicadores químicos, vapor e ETO, indicando que atendem aos dois processos, na verdade atendem somente à Norma dos papeis de Vapor, NB 14990-2. Considerando que o processo de esterilização por vapor exige uma permeância alta para a passagem das moléculas de água, não suportando o uso de papeis de poros fechados, é comum que as embalagens ditas de uso misto, privilegiem as características dos papeis de vapor. Do ponto de vista aplicação no processo, o usuário hospitalar está bem servido, mas essa realidade é justificada em países latino Americanos ou Europeus, onde os métodos de esterilização por ETO e Vapor estão normalmente instalados nos hospitais. No Brasil, onde há uma grande distinção entre Industriais e esterilizadoras usando ETO e de outro lado hospitais usando Vapor, sua utilização não se justificaria pois o tempo de vida para o uso industrial fica extremamente prejudicada.
CONCLUSÃO
Os papeis corretos utilizados para ETO podem dar ao produto realmente uma barreira bacteriana maior. A determinação do prazo de validade depende não só mas também da barreira oferecida por tais papeis. A quebra de barreira, normalmente salvaguardada pela frase “estéril enquanto a embalagem não tiver sido molhada ou violada” precisa também estar assegurada por uma qualificação da embalagem de acordo com o método ao qual se destina.
NBR 14990-3:2010 – Parte 3: Papel grau cirúrgico para fabricação de embalagens para esterilização por processos de baixa temperatura
NBR 14990-2 : 2010 – Parte 2: Papel grau cirúrgico para fabricação de embalagens para esterilização a vapor saturado sob pressão
Quer saber se está usando o papel correto. Veja as especificações, compare o diâmetro dos poros e a permeância. É fácil oferecer e exigir o correto!!!!
Roberto Pereira – Engenheiro graduado pela FESP, Membro participante dos comitês ABNT para Embalagens de Esterilização e Monitores de Processo.